Stand-alone programs, written and maintained by other groups, that generate
complex input for LAMMPS (e.g. force-field fitting), drive LAMMPS as an MD
engine, or otherwise work alongside it. Want to add one? Drop a file in
content/ecosystem/tools/ (or email the developers).
External Tools
PAPRECA
Parallel hybrid off-lattice kMC/MD simulator, with LAMMPS as the MD engine.
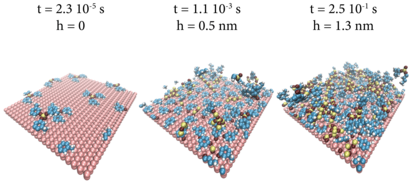
PAPRECA (PArallel PREdefined CAtalog; developer: Stavros Ntioudis, Imperial College London) is a hybrid off-lattice kinetic Monte Carlo / molecular dynamics (kMC/MD) framework. It couples a parallel off-lattice kMC solver with LAMMPS (used as an MD engine library) to resolve both high-activation-energy processes (e.g. adsorption) and high-frequency events (e.g. atomic vibrations). It supports predefined off-lattice events such as reactions, adsorption/desorption, and diffusion hops, and — like LAMMPS — is driven by input files, so no C++ experience is required. Example applications include adsorption/desorption on catalytic surfaces, amorphous thin films, and self-diffusion of gases or solids.
References: JOSS 9(98), 6714 (2024) · Comput. Mater. Sci. 229, 112421 (2023) · tutorials
ELBA-LAMMPS
Tools for simulating the ELBA coarse-grain (lipid/water) model in LAMMPS.
ELBA-LAMMPS (author: Mario Orsi) is a toolkit that assists LAMMPS users in simulating the ELBA coarse-grain model (Orsi & Essex, PLoS One 6: e28637, 2011). It includes installation guidelines, simulation examples, analysis scripts, and tools for visualization with VMD.
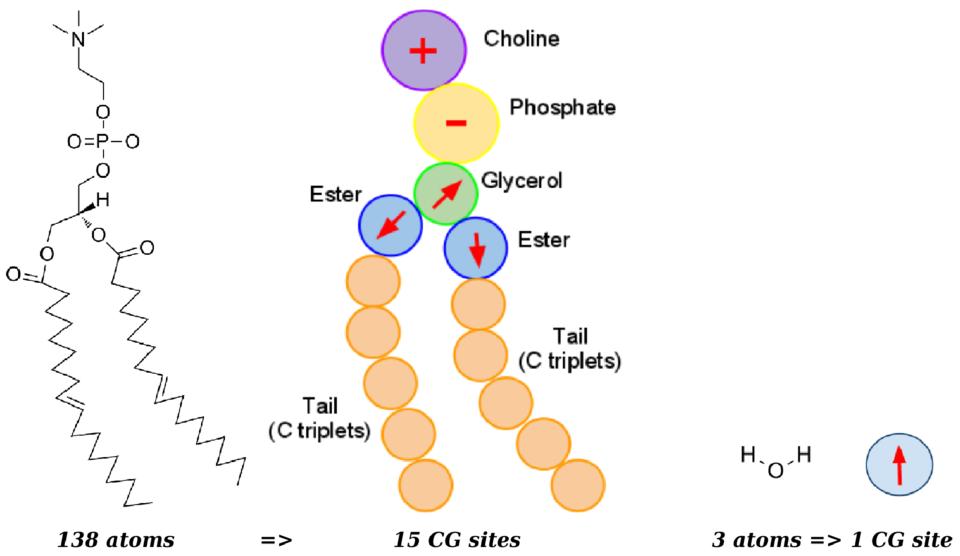
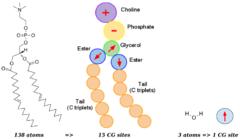
ELBA coarse-grain model of a DOPC phospholipid (left) and a water molecule (right): coarse-grain electrostatics are represented by positive/negative point charges and point dipoles (arrows). Click the image for a larger version.
EPSL
Create and test potential files for pair_style table.
EPSL (easy-pair-table-lammps; author: Luke K. Davis, UCL) is a Python tool to create any pair potential for use with LAMMPS' pair_style table, and provides an automatic check against the pair_write output.
LIGGGHTS
Discrete-element particle simulator for granular materials, built on the LAMMPS engine.
LIGGGHTS (LAMMPS Improved for General Granular and Granular Heat Transfer Simulations; author: Christoph Kloss, JKU / DCS Computing / CFDEM project) is a standalone code focused on granular material modeling that uses LAMMPS internally as a particle engine. On top of the LAMMPS GRANULAR features it adds rewritten contact formulations (including macroscopic particle cohesion), import and handling of triangular CAD meshes, a moving-mesh feature, improved particle insertion, and a model for heat generation and conduction between particles in contact. It stays backward-compatible with LAMMPS, so all LAMMPS features remain available.
PLUMED
Free-energy calculations (metadynamics, umbrella sampling, steered MD) via a patch.
PLUMED is a plugin for free-energy calculations in molecular systems that can be interfaced with many popular molecular dynamics codes through a simple patching procedure. It supports metadynamics with a wide variety of order parameters, combined parallel tempering and metadynamics, bias-exchange metadynamics, umbrella sampling, and steered MD, among many other features, with support for LAMMPS, Quantum-ESPRESSO, NAMD, GROMACS, and more.
VOTCA
Systematic coarse-graining (Boltzmann inversion, force matching) and MD-data analysis.
VOTCA is a set of tools for creating coarse-grained models, simulating charge transport, and more. Its coarse-graining toolkit (VOTCA-CSG) includes Boltzmann inversion for bonded potentials, iterative Boltzmann inversion, inverse Monte Carlo, and force matching, with support for processing LAMMPS dump files.
GARFfield
Multi-objective force-field optimization framework that uses LAMMPS.
GARFfield (author: Andres Jaramillo-Botero, Caltech) is a multi-platform, multi-objective parallel hybrid genetic-algorithm / conjugate-gradient force-field optimization framework. It enables first-principles-based force-field parameter optimization from quantum-mechanical and phenomenological data sets, and supports multiple reactive and non-reactive force fields through LAMMPS — it is particularly suited to developing reactive force fields such as ReaxFF and eFF-ECP. It can produce Pareto-optimal parameters with hill-climbing, fixed/random training-set weights, and deterministic conjugate-gradient refinement near minima. See A. Jaramillo-Botero, S. Naserifar, W. A. Goddard III, J. Chem. Theory Comput. 10(4), 1426–1439 (2014), doi:10.1021/ct5001044.
Jazz
Python wrapper on LAMMPS for vibrational mode lifetimes in solids.
Jazz is a physics research code for calculating the lifetimes of vibrational modes in solids, implemented as a Python wrapper for LAMMPS, which provides the energies and forces for any interatomic potential available in LAMMPS. The anharmonic character of the normal modes is computed via the Monte Carlo-based moments approximation (M. Daw and co-workers). Lead: Prof. Murray Daw (Clemson); contributors: Ted Dickel, Yang Gao, Hengjia Wang. Distributed as open-source software.
USPEX
Universal crystal-structure predictor; interfaces with LAMMPS and many DFT codes.
USPEX enables crystal-structure prediction at arbitrary pressure–temperature conditions given just the chemical composition, with a high success rate. It can also find low-energy metastable phases, stable structures of nanoparticles, surface reconstructions, and molecular packings, and search for materials with desired mechanical or electronic properties. Based on an efficient evolutionary algorithm (with alternatives such as random sampling, metadynamics, and corrected PSO), USPEX is interfaced with many DFT and classical codes, including VASP, SIESTA, GULP, Quantum Espresso, CP2K, CASTEP, and LAMMPS.
LAMMPS extension for VS Code
Visual Studio Code extension for LAMMPS input files.
An extension of the Visual Studio Code editor for LAMMPS input files, available through the editor’s extension system, the VS Marketplace, or GitHub. Features include keyword/syntax highlighting, embedded offline documentation, autocompletion, hover information, a task provider for running LAMMPS in the editor terminal, input-file checks, and a dashboard for interactive plots of logs and atomic dumps. See the documentation.