This page points to software tools, data repositories, and other resources that
work well alongside LAMMPS — whether freeware or commercial. They extend the
range of problems LAMMPS can model and make it easier to use. Want to add one?
Drop a file in content/ecosystem/prepost/ (or email the developers).
Pre/Post Processing Tools
GUI, Analysis & Workflow
Commercial and free software that wraps LAMMPS to build models, run simulations, and analyze results.
Materials Design (MedeA®)
MedeA® — commercial atomistic modeling environment with builders and analysis for LAMMPS.
Materials Design, Inc. develops MedeA®, an atomistic simulation and modeling environment that provides productivity, model-building, and analysis tools for use with LAMMPS. MedeA® simplifies LAMMPS simulations with flowcharts that assemble complex protocols from discrete LAMMPS stages, which can be shared, edited, and reused. Atomistic models can be constructed with the collection of MedeA® builders, and validated methods predict mechanical properties, elastic constants, diffusivity, transport properties, and cohesive energies from LAMMPS simulations. MedeA® also manages force fields for organic, inorganic, and metallic systems.
OVITO
Visualization and analysis for atomistic, MC, and other particle-based simulation data.
OVITO is a visualization and analysis software for output data generated in molecular dynamics, atomistic Monte-Carlo, and other particle-based simulations. It is particularly well interfaced with LAMMPS, since some of the OVITO developers are frequent LAMMPS users. The OVITO Pro version can also load the LAMMPS shared library to create and run LAMMPS simulations directly from OVITO, loading the data without intermediate files.
Scienomics (MAPS)
MAPS platform — commercial build/simulate/analyze environment with LAMMPS plugin and force fields.
Scienomics has developed an interface to LAMMPS as part of their Materials and Processes Simulations (MAPS) platform, which lets both novice and expert users quickly create LAMMPS input files for atomistic and DPD simulations, with full phone and email support. The LAMMPS plugin within MAPS also lets users visualize and analyze LAMMPS output, and MAPS offers a number of atomistic and coarse-grained force fields plus builder tools for creating input geometries. MAPS implements a complete Build–Simulate–Analyze workflow across an array of simulation engines including LAMMPS.
Atomify
Run LAMMPS in the web browser with real-time visualization and plotting — no installation.
Atomify is a web-based application that runs LAMMPS simulations purely in the browser, with real-time visualization and plotting of thermodynamic quantities. It ships with several example simulations and lets you analyze them in a browser-based Jupyter notebook. Because it requires no installation, it is an excellent choice for new LAMMPS users and for teaching environments.
atomman
NIST Python package to prepare, run, and analyze atomistic systems and defects with LAMMPS.
atomman is open-source software developed by Lucas Hale at NIST: a Python package for interacting with large-scale atomic systems that lets users prepare, run, and analyze MD simulations entirely from Python. It supports efficient calculations on millions of atoms with many per-atom properties, generation of defects (point defects, dislocation monopoles), built-in defect analysis (slip vector, Nye tensor), and direct calls to LAMMPS from Python. It converts to/from ASE and PyMatGen, loads CIF/POSCAR and LAMMPS atom/dump files, includes unit-conversion tools, and runs on Linux, Windows, and macOS.
LAMMPS-GUI
Cross-platform GUI editor that runs, monitors, and visualizes LAMMPS via the library interface.
LAMMPS-GUI is a cross-platform graphical text editor for LAMMPS input files with
syntax highlighting, auto-completion, inline help, and indentation support.
Written in C++ with the Qt framework, it calls LAMMPS directly through the
library interface instead of launching an external executable, so it can display
information while LAMMPS runs and show visualizations created by the dump image
command. It is well suited to teaching beginners with consistent behavior across
Linux, macOS, and Windows, and is integrated with a collection of LAMMPS
tutorials. It is written and maintained by Axel Kohlmeyer.
LOOS
Lightweight Object-Oriented Structure analysis library — ~150 tools for MD trajectories.
LOOS (Lightweight Object-Oriented Structure analysis library) is a package-agnostic, open-source package for analyzing molecular dynamics simulations that runs on all major Linux distributions and macOS. It ships with roughly 150 pre-packaged analysis tools, from standard tasks (trajectory manipulation, principal component analysis) to novel ones (assessing convergence, measuring membrane properties), and is designed for rapid development of new analysis tools, particularly via its Python wrappers. It is available on GitHub.
LUNAR
LAMMPS Utility for Network Analysis and Reactivity — Python pre/post-processing toolkit.
LUNAR (LAMMPS Utility for Network Analysis and Reactivity) is a standalone Python (3.7+) toolkit that supplements LAMMPS, focused on pre- and post-processing of LAMMPS inputs and outputs with an emphasis on structure–property relationships for ICME process modeling of polymers — though many of its codes are useful outside process modeling. It was written by Josh Kemppainen (Michigan Technological University, advisor Dr. Gregory M. Odegard) and is maintained by Josh Kemppainen and Dr. Jacob R. Gissinger. All source is provided so users can modify it.
When using LUNAR, please cite “LUNAR: Automated Input Generation and Analysis for Reactive LAMMPS Simulations”, https://doi.org/10.1021/acs.jcim.4c00730.
freud
Python analysis library for MD/MC: RDFs, correlation functions, order parameters, clustering.
freud is a Python library developed and supported by Sharon Glotzer’s group (University of Michigan) that provides a simple, flexible, powerful set of tools for analyzing trajectories from molecular dynamics or Monte Carlo simulations. High-performance, parallelized C++ computes standard tools such as radial distribution functions, correlation functions, order parameters, and clusters, as well as original methods including potentials of mean force and torque (PMFTs) and local-environment matching. freud supports many input formats and outputs NumPy arrays, integrating with the scientific Python ecosystem.
- Tutorial: g(r) analysis
- Documentation: https://freud.readthedocs.io
MDAnalysis
Object-oriented Python library to read and analyze MD trajectories via NumPy arrays.
MDAnalysis is an object-oriented Python library to analyze trajectories from molecular dynamics simulations. It reads particle-based trajectories (including single coordinate frames such as biomolecules in PDB format) and exposes atomic coordinates as NumPy arrays, providing a flexible and relatively fast framework for complex analysis tasks. It implements powerful atom-selection commands, and trajectories can be manipulated (for example, fit to a reference structure) and written out.
MDANSE
Compute neutron-scattering observables (INS/QENS) from MD trajectories.
MDANSE (Molecular Dynamics Analysis of Neutron Scattering Experiments) helps predict neutron observables from MD trajectories. Inelastic and quasi-elastic neutron scattering (INS and QENS) probe molecular dynamics in materials, and MDANSE implements the operations needed to compare simulations with experiment — velocity auto- and cross-correlation functions, Fourier transforms, and convolutions with instrument parameters. It interfaces with more than ten MD codes, including LAMMPS, CASTEP, VASP, Gromacs, CHARMM, and DFTB. The package is being modernized and is available on GitHub and PyPI.
mdapy
Cross-platform, parallel Python library for analyzing MD trajectories (CPU/GPU).
mdapy (Molecular Dynamics Analysis with Python) offers a wide range of flexible, user-friendly tools for analyzing atomic trajectories from MD simulations across Windows, Linux, and macOS. It uses the TaiChi project to accelerate pure Python toward C++ performance and is optimized for parallel processing on multicore CPUs and GPUs. mdapy handles LAMMPS DUMP and DATA files, VASP POSCAR, universal XYZ, and CIF formats, and stores all data as NumPy ndarrays for seamless integration with the scientific Python ecosystem.
- Documentation: https://mdapy.readthedocs.io/
- Publication: https://doi.org/10.1016/j.cpc.2023.108764
MolTwister
Command-line package to construct molecular systems and analyze MD trajectories, with a 3D view.
MolTwister is an open-source package that aids in constructing molecular systems for MD simulations, but whose primary purpose is a collection of analysis tools for post-processing trajectories and data — density profiles, velocity autocorrelation functions, radial distribution functions, dihedral distributions, and more. It runs as an extension of the command line: commands it does not recognize are passed through to the regular shell, and a 3D view window displays the molecular system being edited or created. See the documentation.
PyLAT
Python LAMMPS Analysis Tools for post-processing LAMMPS output (Maginn group).
PyLAT (Python LAMMPS Analysis Tools) is Python software developed in the group of Ed Maginn at Notre Dame for post-processing output from LAMMPS simulations. Details are in this publication.
TRAVIS
Trajectory Analyzer and Visualizer — open-source tool collecting many MC/MD analyses.
TRAVIS (“TRajectory Analyzer and VISualizer”) is a free program package for analyzing and visualizing Monte Carlo and molecular dynamics trajectories. Its aim is to collect as many analyses as possible in one program, making it unnecessary to use many different tools to evaluate simulations. TRAVIS is written in C++, is open-source freeware under the GPLv3, is platform-independent with no external libraries, and is easy to install and use.
Molecular Builders
Tools that build or convert molecular inputs (topologies, coordinates, force-field coefficients) for LAMMPS.
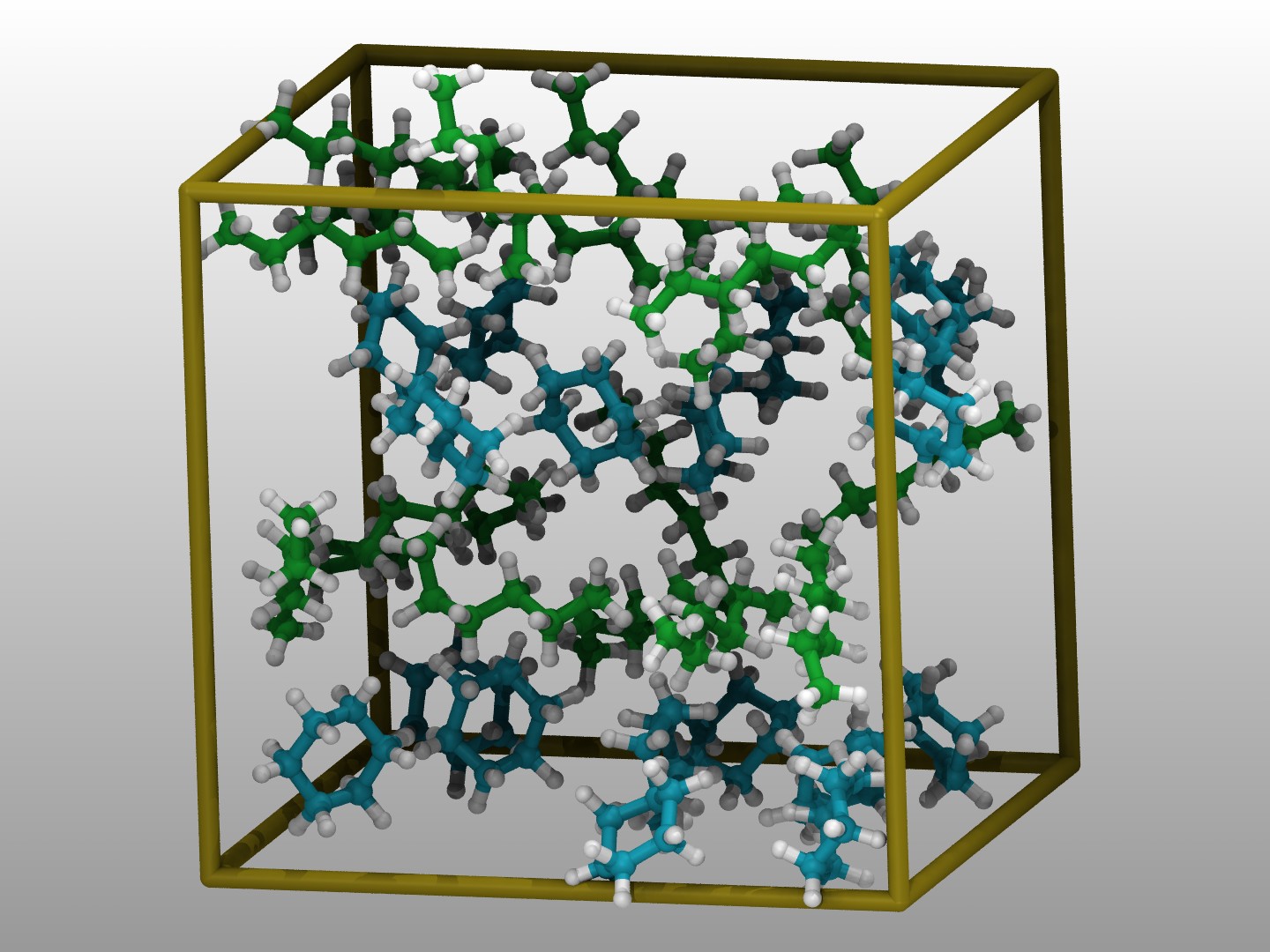
Enhanced Monte Carlo (EMC)
Build and manipulate input structures from SMILES for many force fields; exports to LAMMPS.

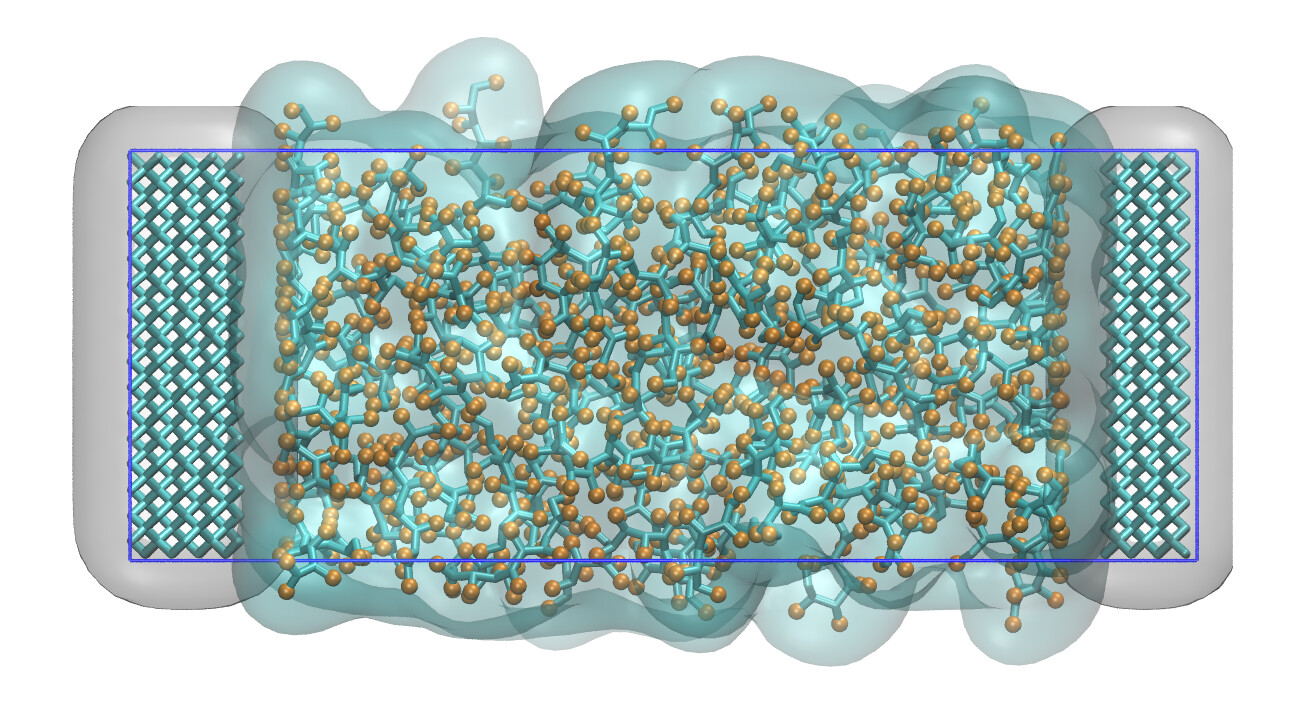
Developed and maintained by Pieter J. in ’t Veld (BASF). Enhanced Monte Carlo (EMC) provides an environment for creating and manipulating input structures for particle simulations using COMPASS, CHARMM, OPLS, Martini, DPD, or colloidal force fields. A scripting language manages access to its functionality: manipulation of molecular or coarse-grained structures through SMILES strings, typing those structures for selected force fields, and building conformations using Monte Carlo principles to unoverlap atoms. EMC provides output ports to LAMMPS, PDB, and XYZ formats; compiled versions for Linux, macOS, and Windows are available. EMC has a user forum on MatSci.org.
Examples of systems built with EMC (click for a larger image):

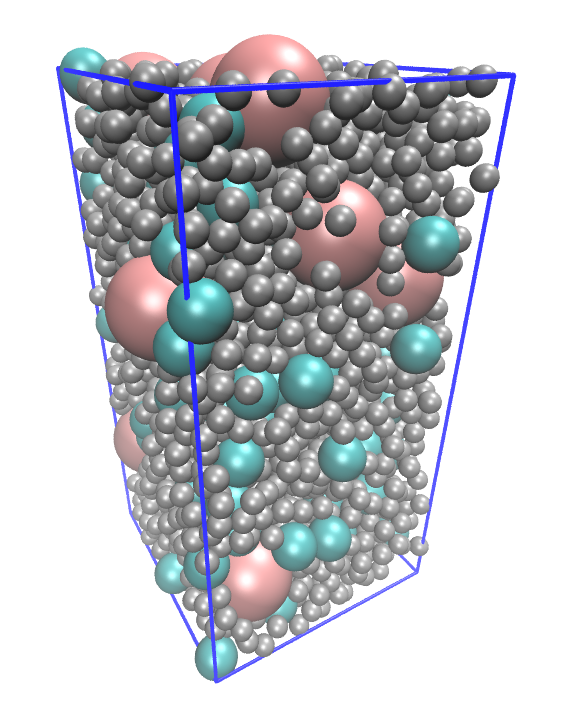
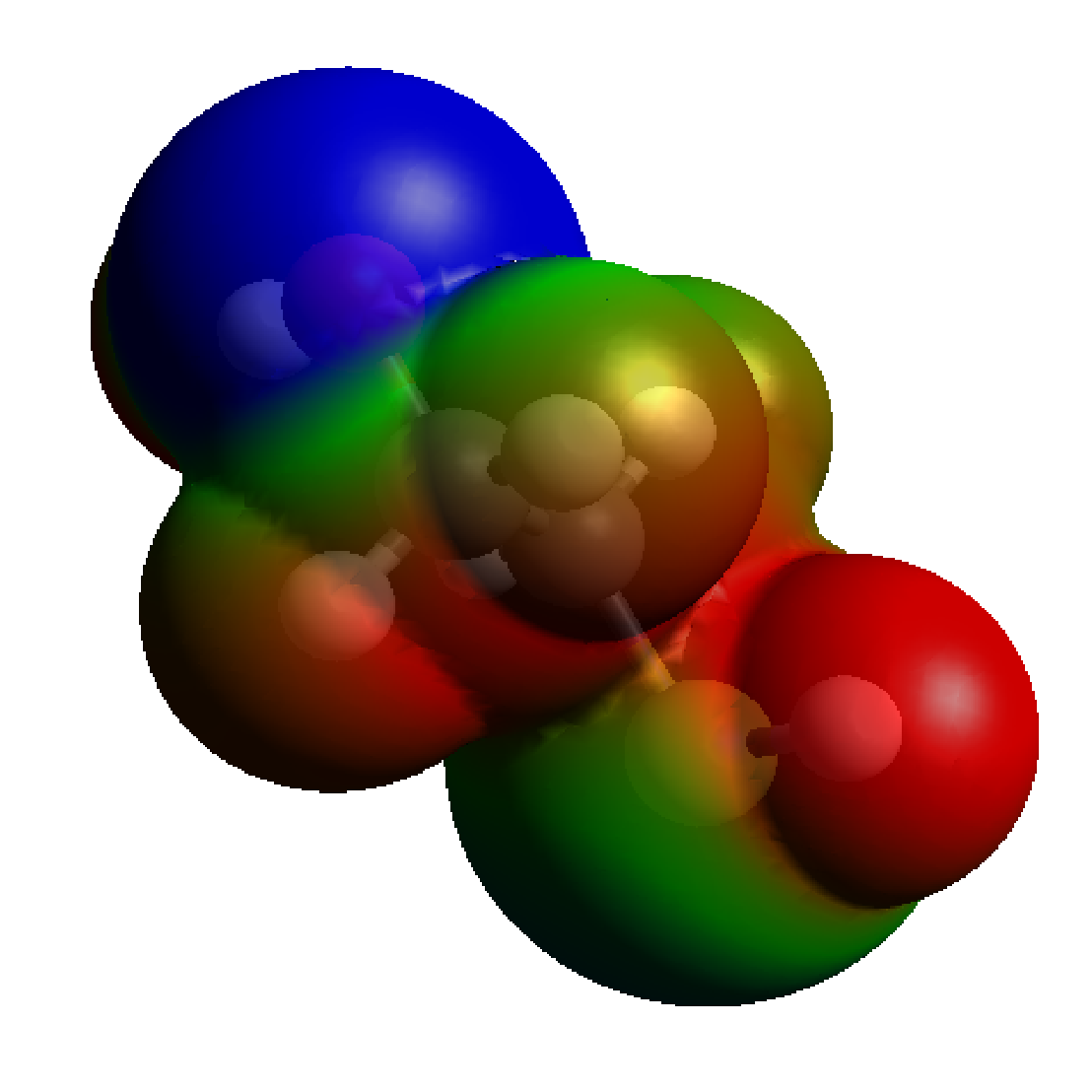
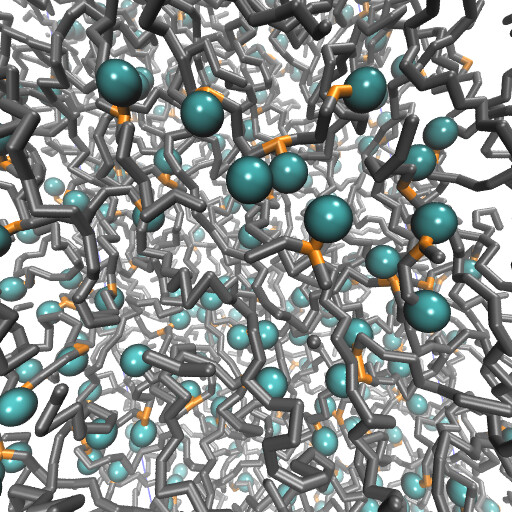
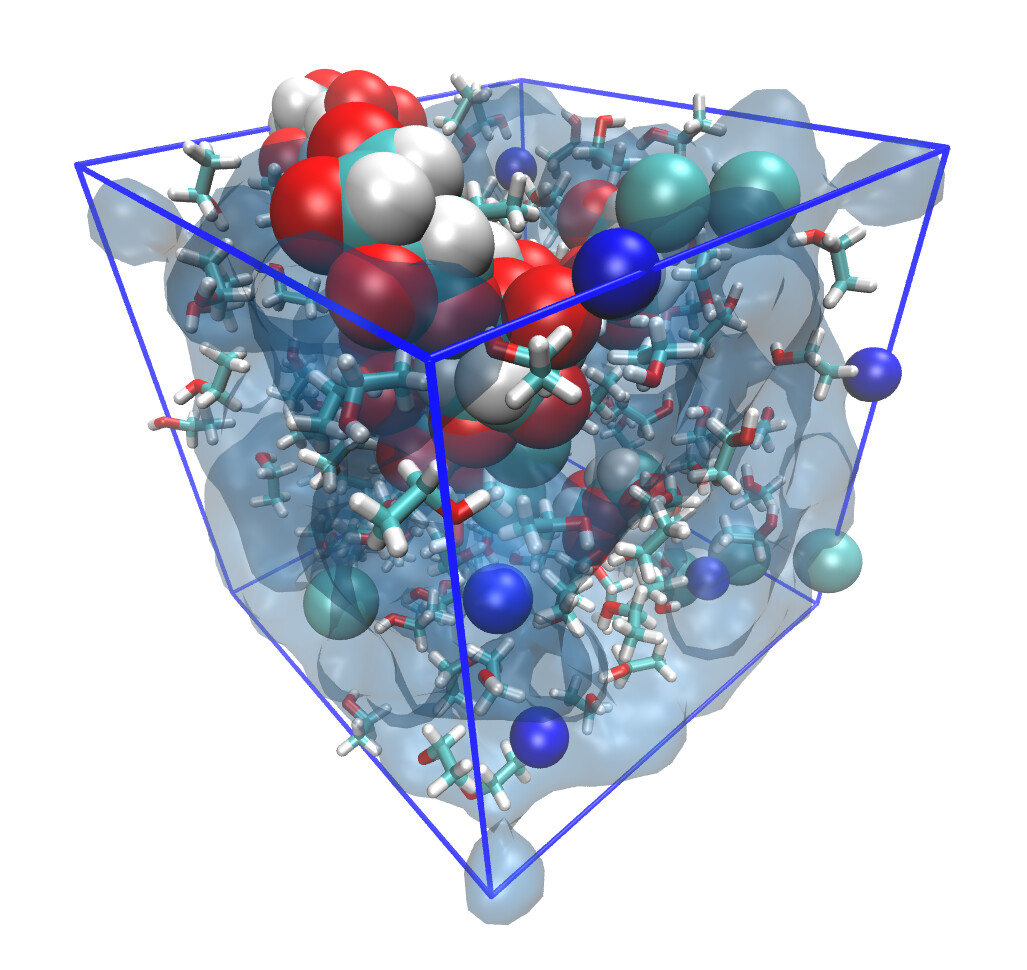

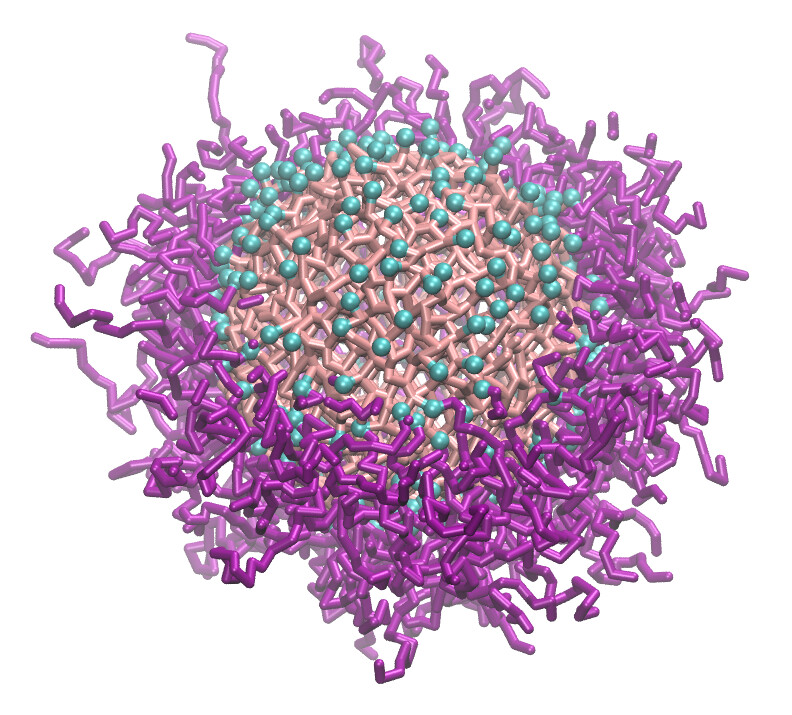

Moltemplate
Build LAMMPS data and input files from reusable, nestable molecule templates.
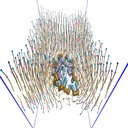
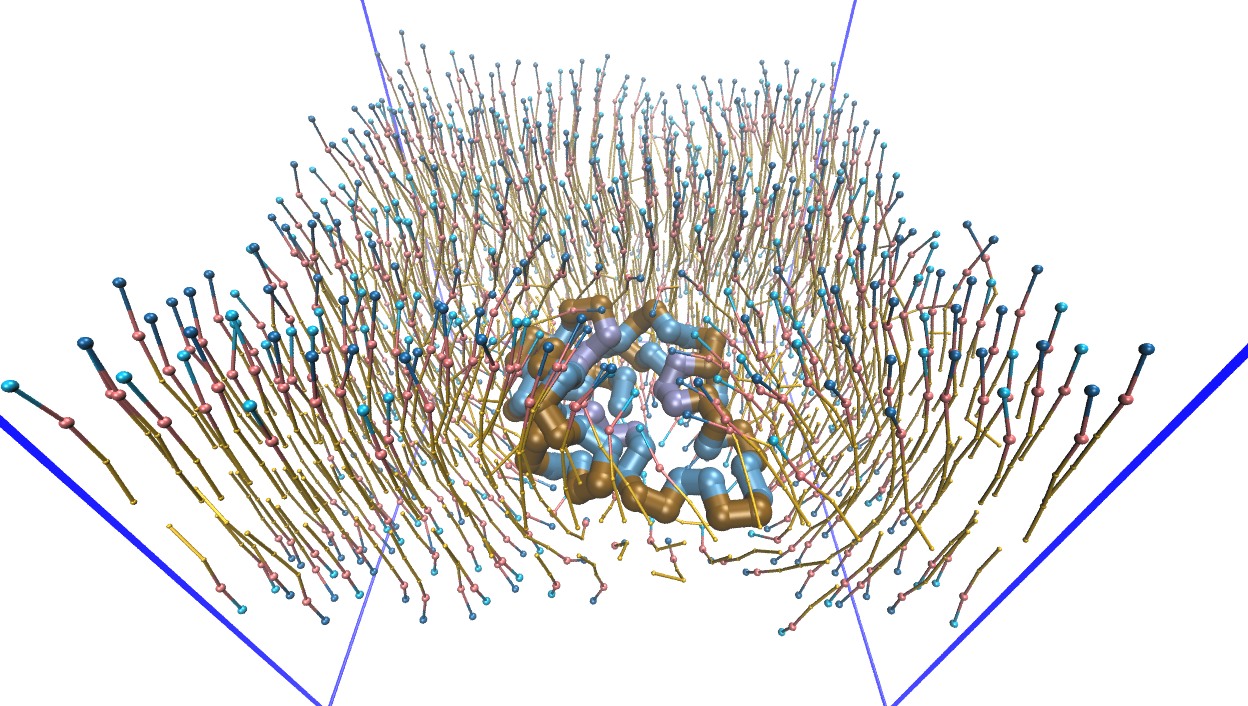
Moltemplate, developed and maintained by Andrew Jewett (UCSB), is distributed
with LAMMPS in the tools/moltemplate directory. It was designed for building
coarse-grained biomolecular models and can create both LAMMPS data files
(geometry and topology) and LAMMPS input scripts (force fields, fixes, groups),
giving users access to all of the force fields available in LAMMPS. Molecules are
written in a compact, readable template format (.LT), can be shared, and can be
copied, combined, and nested to build larger molecules.
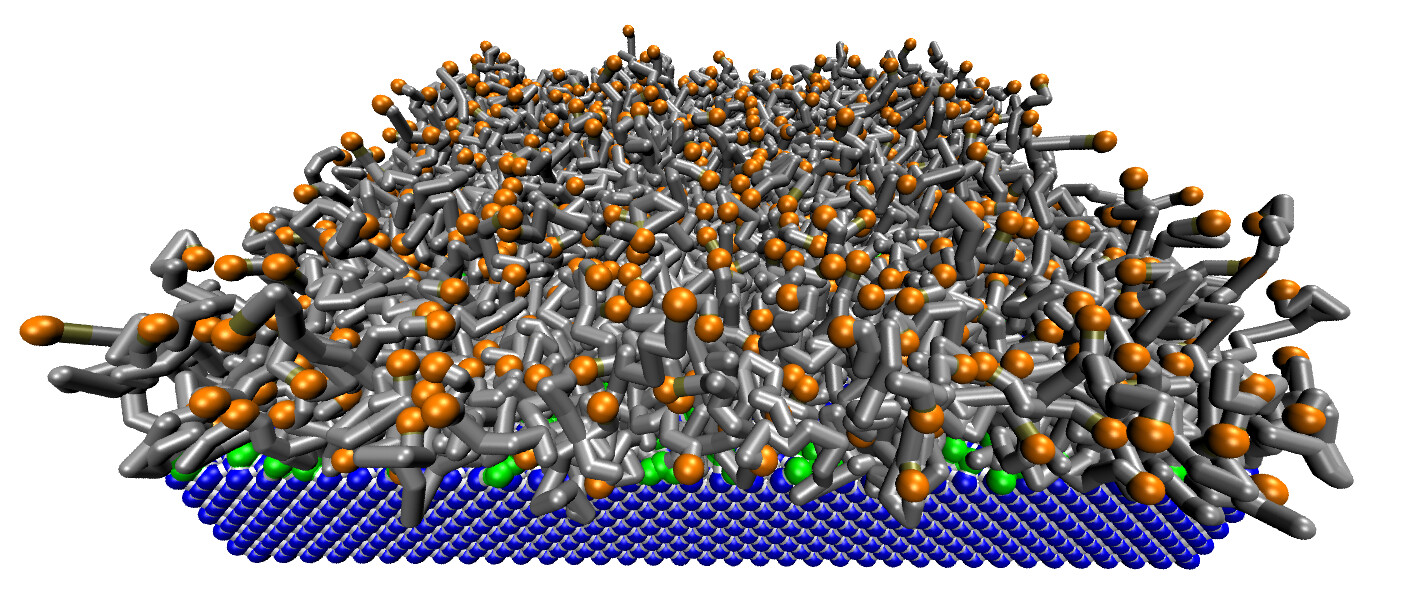
Examples of systems built with Moltemplate (click for a larger image):

VMD TopoTools
VMD/Tcl molecule builder that reads, writes, replicates, and merges LAMMPS data files.

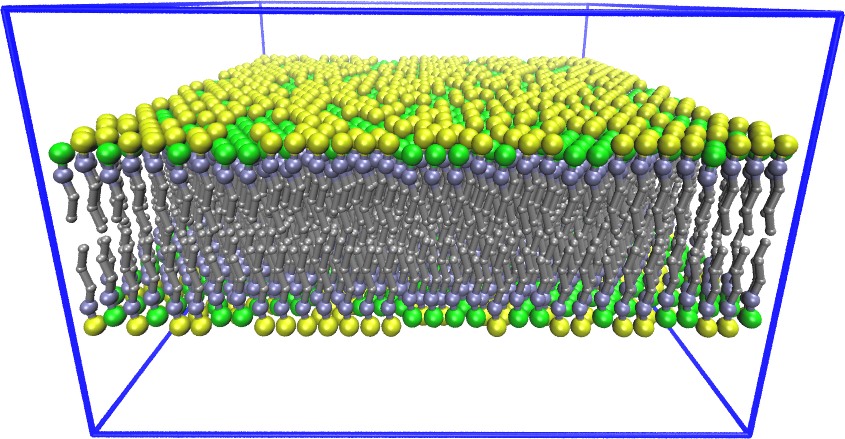
Developed and maintained by Axel Kohlmeyer (Temple U). TopoTools is a molecule builder that leverages VMD and Tcl to create LAMMPS data files and convert them to and from other formats. It has two components: a middleware script that extracts and manipulates topology information, and several high-level applications built on top of it (for example, to read/write data files and replicate and merge systems). Together with VMD, TopoTools can infer topology from PDB and PSF files and atom-pair distances, and solvate a protein.
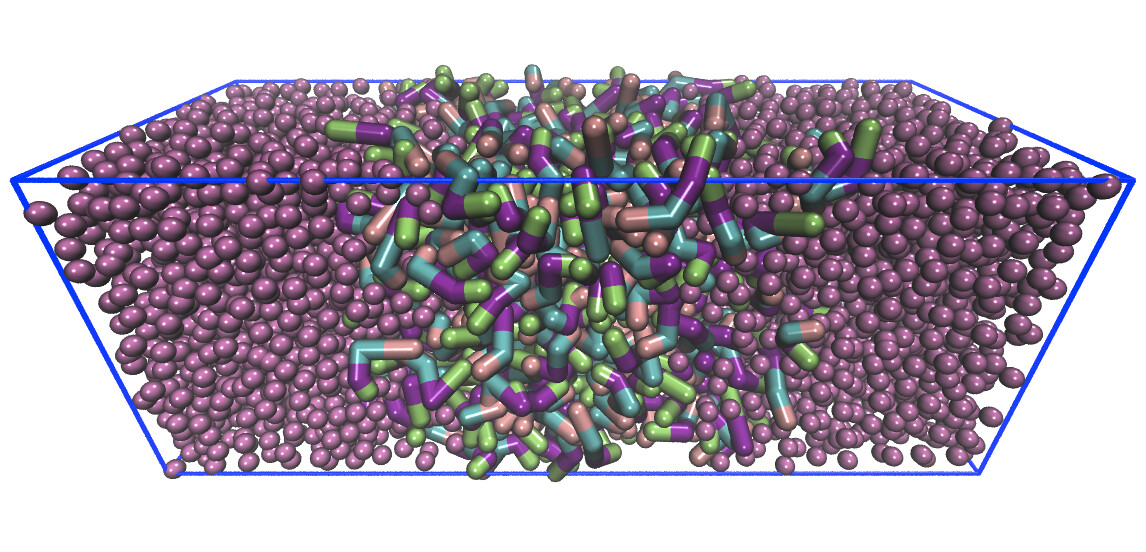
Examples of systems built with TopoTools (click for a larger image):


InterMol
Convert molecular dynamics input between Desmond, Gromacs, LAMMPS, AMBER, and CHARMM.
InterMol is written in Python and performs Desmond ⇄ Gromacs ⇄ LAMMPS conversions natively. AMBER → X is carried out by converting AMBER to GROMACS and then to other programs using ParmEd; AMBER → CHARMM is done by ParmEd directly.
AMBER2LAMMPS
Python utility to convert AMBER molecular dynamics files to LAMMPS data format.
AMBER2LAMMPS is a Python utility to convert AMBER molecular dynamics files to the LAMMPS data format, with an enhanced command-line interface and error handling. It can also be loaded as a Python module to embed the conversion in more complex workflows, and it uses ParmEd.
Avogadro
Advanced open-source molecule editor and visualizer with a plugin architecture.
Avogadro is an advanced molecule editor and visualizer designed for cross-platform use in computational chemistry, molecular modeling, bioinformatics, materials science, and related areas. It offers flexible, high-quality rendering and a powerful plugin architecture, and is developed and maintained as an open-source project.
Packmol
Pack molecules into defined regions to create starting points for MD simulations.
Packmol creates an initial configuration for molecular dynamics simulations by packing molecules into defined regions of space. The packing guarantees that short-range repulsive interactions do not disrupt the simulations. It is developed and maintained as an open-source project.
Atomsk
Create, manipulate, and convert atomic systems across many file formats, including LAMMPS.
Atomsk creates, manipulates, and converts atomic systems. It supports many file formats — among them LAMMPS, VASP, Quantum Espresso, IMD, DL_POLY, AtomEye CFG, and xCrySDen XSF — making it easy to convert files for ab initio calculations, classical-potential simulations, or visualization. Atomsk can also perform simple transformations of atomic positions such as rotation, deformation, and inserting dislocations. It is developed and maintained as an open-source project.
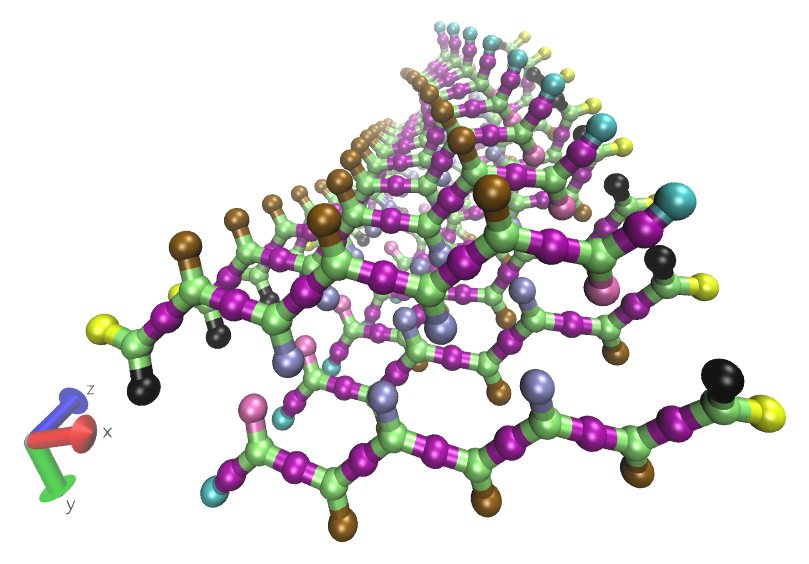
polyGraft
Build initial structures and topologies for polymer-grafted hybrid systems.
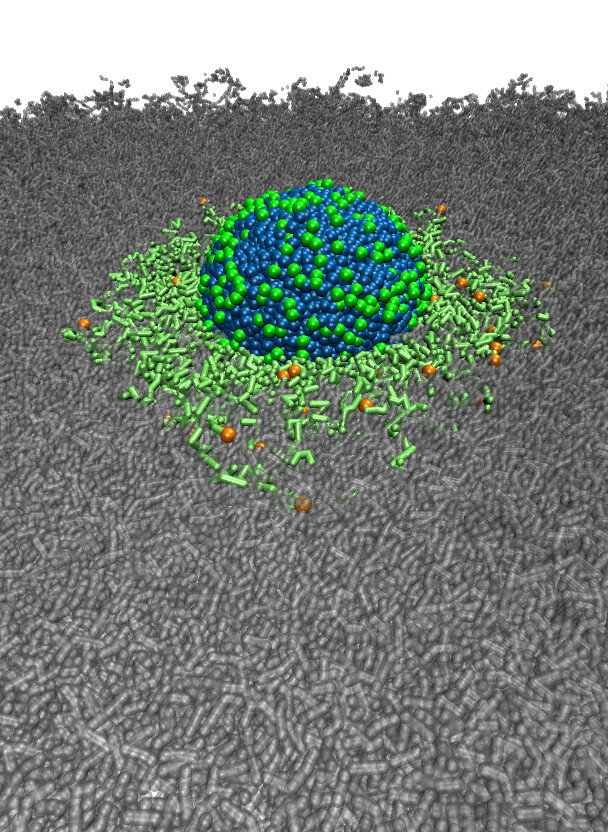
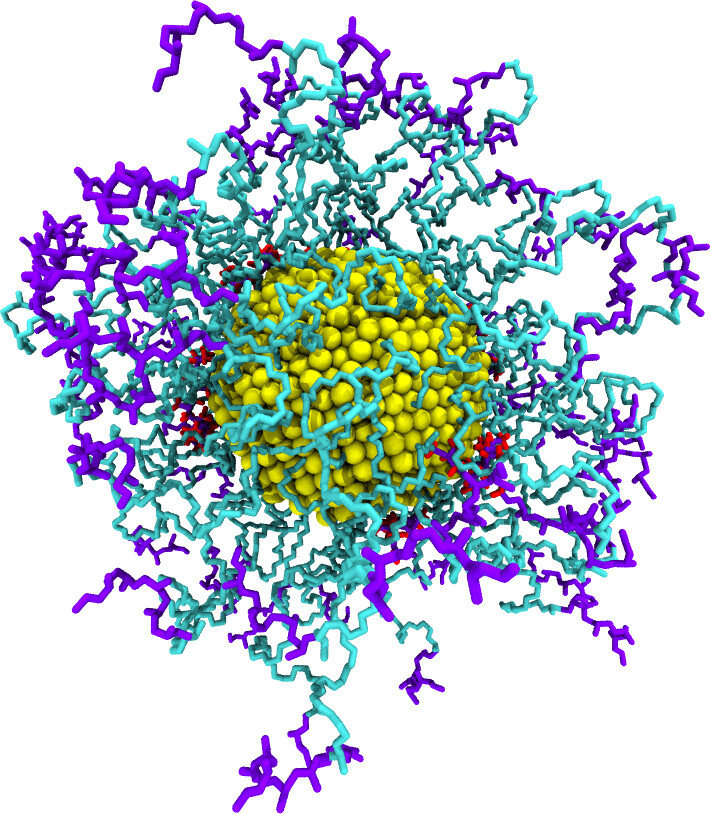
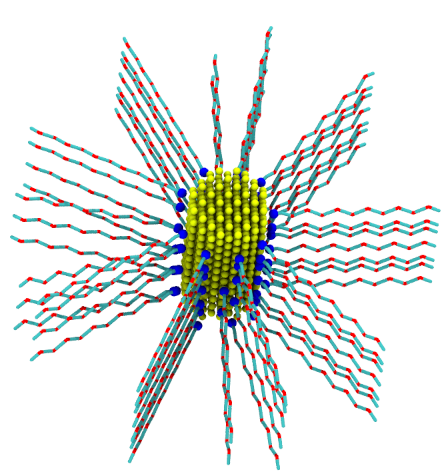
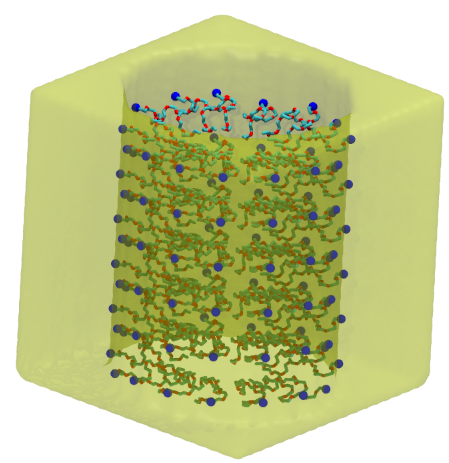
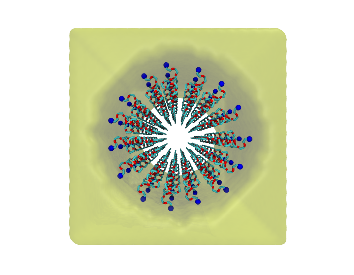
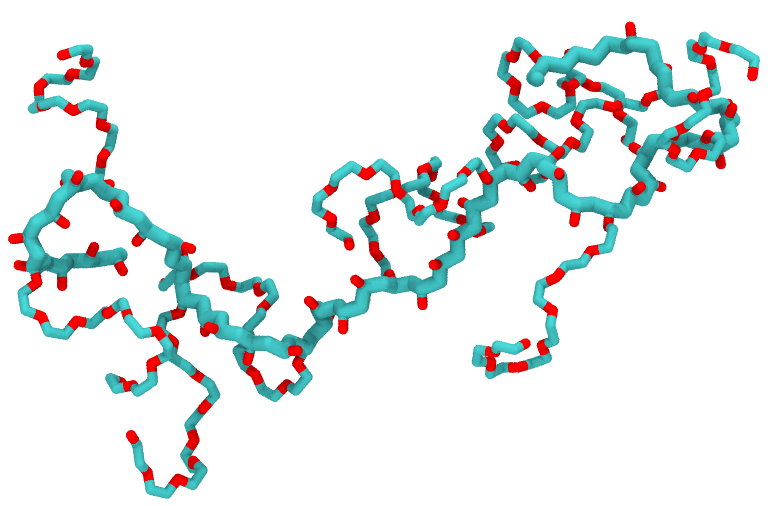
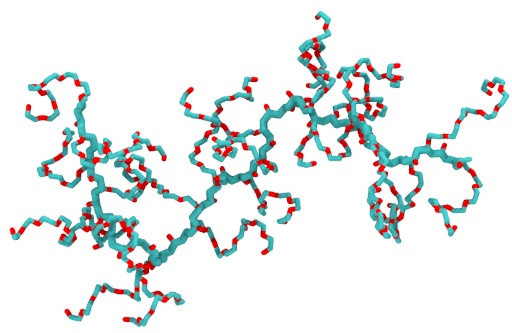
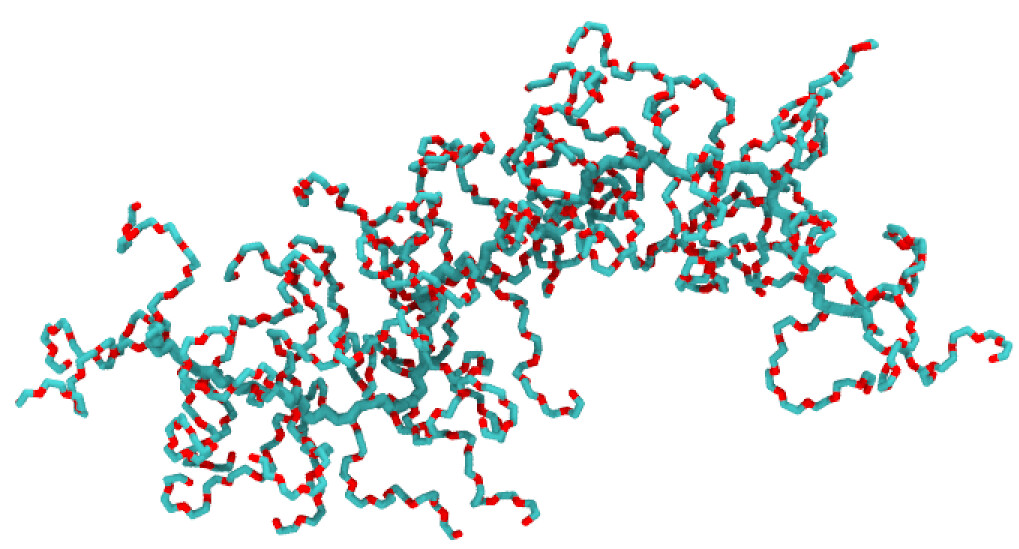
polyGraft is developed and maintained by Guang Chen to help build the initial structure and topology of polymer-grafted hybrid systems. A broad range of polymer-grafted hard or soft nanostructures can be generated — polymer-grafted nanoparticles, nanoslabs (planar brushes), nanorods/nanopores, and bottlebrush polymers — accounting for the chain length and grafting density of the polymer graft and the size and shape of the substrate.
Examples of systems built with polyGraft (click for a larger image):

OCTA / J-OCTA
Soft-matter simulation suite (MD, rheology, SCFT, FEM) with a GUI; commercial J-OCTA adds builders.
OCTA is an open-source software package consisting of simulation engines (molecular dynamics, rheology simulation, self-consistent field theory, finite element method, and more) and a GUI for visualization, simple molecular building, and analysis of soft-matter systems. It also provides an environment for the collaborative use of several simulators (multi-physics and multi-scale simulations). The commercial J-OCTA adds complex molecular building for full-atomistic and coarse-grained MD.
LAMMPS Interface
Generate LAMMPS input files from crystallographic CIF files using the UFF force field.
LAMMPS Interface is a Python program designed as an easy-to-use interface between
crystallographic information files (.cif) and LAMMPS. By default it creates
LAMMPS input files using the UFF force field from CIF files; the CIF files are
expected to be in P1 symmetry.
MoSDeF
Molecular Simulation Design Framework — Python tools to initialize, atom-type, and screen systems.
The Molecular Simulation Design Framework (MoSDeF) is a set of extensible Python tools designed to facilitate the initialization, atom-typing, and screening of soft-matter systems using molecular simulation.
Automated Topology Builder (ATB)
Repository of organic-molecule topologies in LAMMPS-compatible formats (>20,000 molecules).
The ATB project is led by Prof. Alan E. Mark (University of Queensland). It provides topology files for organic molecules in formats compatible with LAMMPS and other molecular dynamics packages; the LAMMPS topology files enable building complex systems with the Moltemplate tool distributed with LAMMPS. The site provides topologies for a wide variety of molecules (>20,000 and growing), searchable by name or chemical formula. On a molecule’s page, choose “Molecular Dynamics (MD) Files” and select “LAMMPS” as the output format; molecules not yet in the database can be submitted for processing.
cif2cell
Generate simulation geometry from CIF files for many codes, including LAMMPS.
CIF2Cell generates the geometrical setup for various electronic-structure and
simulation codes from a CIF (Crystallographic Information Framework) file. It
supports output for many programs, including LAMMPS, ABINIT, ASE, CASTEP, CP2K,
CPMD, CRYSTAL09, Elk, EMTO, Exciting, Fleur, FHI-aims, MOPAC, Quantum Espresso,
RSPt, Siesta, SPR-KKR, and VASP, plus common geometry formats (.coo, .cfg,
.xyz). It was published in Computer Physics Communications 182 (2011)
1183–1186, and can be installed via
pip or
conda-forge.
Geoparticle
Python tool to construct particle-based geometries with smooth surfaces.
Particles of specified geometries are typically created by the lattice command
in LAMMPS, which can produce rough surfaces when the particle spacing is not
small enough — while too small a spacing yields too many particles and higher
cost. A similar issue arises when creating atoms from an external STL file.
Geoparticle aims to easily construct geometries where smooth surfaces are
required. It is hosted on GitHub,
documented on Read the Docs,
and installable via pip
(pip install geoparticle).
Data Sites
Websites with data usable as LAMMPS input, or that archive LAMMPS output.
JARVIS-FF
NIST high-throughput database of force-field calculations on DFT-optimized geometries.

JARVIS for force-fields (JARVIS-FF) is a high-throughput computational database for LAMMPS calculations on density-functional-theory-optimized geometric structures with various force fields / interatomic potentials. Its goal is to provide an easy look-up table for evaluating force fields through a web interface and to enhance data reproducibility. JARVIS-FF is part of the Materials Genome Initiative (MGI) at the National Institute of Standards and Technology (NIST).
NIST Interatomic Potentials Repository
Curated collection of interatomic potentials (force fields) plus tools to evaluate them.
Hosted by the National Institute of Standards and Technology (NIST), the Interatomic Potentials Repository is a curated source of interatomic potentials (force fields), their parameter files, and evaluation tools. It spans many potential classes (EAM, MEAM, ReaxFF, and others) and material systems, with files submitted or vetted by developers and accompanied by the relevant citations; many are provided in formats ready to use with LAMMPS. The site also publishes computed properties — lattice constants, elastic constants, melting temperatures, and more — so users can compare potentials and pick one suited to their problem.
Crystallography Open Database (COD)
Open-access collection of >500,000 crystal structures in CIF format.
The Crystallography Open Database (COD) is an open-access collection of crystal structures of organic, inorganic, metal-organic compounds and minerals (excluding biopolymers). It contains over half a million crystal structures in CIF format, which can be converted into LAMMPS input with tools such as cif2cell or LAMMPS Interface.